
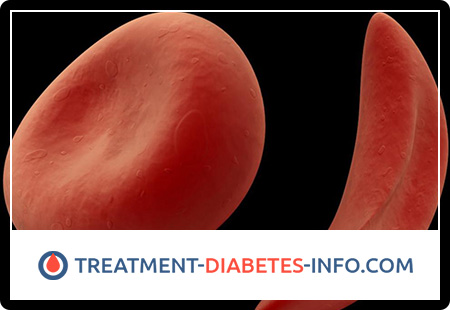
What is Sickle Cell Anemia?
Sickle cell anemia is a hereditary hemoglobinopathy associated with such a violation of the structure of the hemoglobin protein, in which it acquires a special crystalline structure – the so-called hemoglobin S. The red blood cells that carry hemoglobin S instead of normal hemoglobin A under the microscope have a characteristic sickle-shaped (sickle shape) , for which this form of hemoglobinopathy was called sickle cell anemia.
Sickle cell anemia is very common in malaria endemic regions of the world, and sickle cell anemia patients have an increased (although not absolute) congenital resistance to infection with various strains of malarial plasmodium. The sickle-shaped erythrocytes of these patients are also not susceptible to in vitro malaria plasmodium infection.
Causes of Sickle Cell Anemia
Sickle cell anemia is inherited in an autosomal recessive manner. In patients heterozygous for the sickle cell anemia gene, along with sickle-shaped erythrocytes that carry hemoglobin S, normal ones that carry hemoglobin A are also present in the blood. In this case, the disease is less clinically pronounced, it is easier, and sometimes it does not cause symptoms, and sickle-shaped red blood cells detected by chance in a laboratory study of blood. Homozygotes for the sickle cell anemia gene in the blood have only sickle red blood cells carrying hemoglobin S, and the disease is severe.
If both parents carry the sickle cell anemia gene, then their children are 25% likely to have sickle cell anemia and there is a 50% chance to inherit only their predisposition to it. If only one parent has the sickle cell anemia gene, there is absolutely no risk that the child will have sickle cell anemia, although there is a 50% chance that the child will inherit one sickle cell anemia gene.
Pathogenesis during Sickle Cell Anemia
The erythrocytes carrying hemoglobin S have reduced resistance and reduced oxygen transporting ability, therefore, in patients with sickle cell anemia, the destruction of red blood cells in the spleen is increased, their life is shortened, hemolysis is increased, and there are often signs of chronic hypoxia (oxygen deficiency) or chronic ” redistribution of erythrocyte bone marrow growth.
Symptoms of Sickle Cell Anemia
Sickle cell anemia is clinically characterized by symptoms caused, on the one hand, by vascular thrombosis of various organs with sickle-shaped red blood cells, and on the other, by hemolytic anemia. The severity of anemia depends on the concentration of HbS in the red blood cell: the larger it is, the brighter and more severe the symptoms. In addition, other pathological hemoglobins may be present in red blood cells: HbF, HbD, HBS, etc. Sometimes sickle cell anemia is combined with thalassemia, while clinical manifestations may decrease or, on the contrary, increase.
In the initial period of the disease, the bone marrow system is mainly affected: a swelling appears, as well as pain due to thrombosis of the vessels supplying the joint and bone. Aseptic necrosis of the femoral head is possible with the subsequent attachment of infection and osteomyelitis. Hemolytic crises usually develop after infections, are regenerative or hyporegenerative in nature and are the main cause of death of these patients. In rare cases, sequestration crisis is observed due to deposition of blood in the spleen and liver, which is expressed by pain abdominal syndrome due to the rapid increase in these organs and is accompanied by collapse; while hemolysis may be absent, pulmonary infarction occurs due to impaired microcirculation at the level of the pulmonary vessels.
In the second period, a constant symptom is hemolytic anemia. Bone marrow hyperplasia developing in the tubular bones (active hemopoiesis is performed as a compensatory reaction to hemolysis) is accompanied by characteristic changes in the skeleton: thin limbs, a curved spine, a tower skull with bulges in the forehead and parietal bones. Hepato- and splenomegaly develop due to the activation of erythropoiesis in them, as well as secondary hemochromatosis and thrombosis; in some patients, gallstone disease is formed. Hemosiderosis of the heart muscle leads to heart failure, and hemosiderosis of the liver, pancreas leads to cirrhosis and diabetes mellitus.
Renal vascular thrombosis occurs with hematuria and subsequent renal failure. Neurological symptoms are caused by stroke, cranial nerve palsy, etc. Trophic ulcers on the lower extremities are characteristic. Most patients with severe sickle cell anemia die within 5 years, and survivors of this period enter the third period, which is characterized by signs of mild hemolytic anemia. Their spleen is usually not palpable, since repeated heart attacks lead to its wrinkling – autosplenectomy. The liver remains enlarged, unevenly compacted, and frequent infections often take a septic course.
Hematologic changes. Hemoglobin concentration decreases (<80 g / l) and averages 50 g / l, especially during hemolytic crisis. Normochromic anemia, regenerative; reticulocytosis – 5-15%. Red blood cells with Jolly’s bodies are found. The number of leukocytes during the crisis is increased to 20 × 109 / L. In the bone marrow, hyperplasia of the erythroid germ is observed. To detect the sickle shape of red blood cells, a special test is carried out: a drop of blood is covered with glass, sealed, for which the edges of the glass are smeared with petroleum jelly; after a few minutes, the partial pressure of oxygen in a drop of blood under the glass decreases and the red blood cells take a sickle shape. Hemoglobin electrophoresis is more informative: with sickle cell anemia in homozygotes, the bulk is HbS, HbA is absent, the HbF content is increased; in heterozygotes, electrophoresis reveals HBA along with HbS. In the blood, the content of the free fraction of bilirubin is increased, the content of serum iron is increased; osmotic resistance of red blood cells is increased.
Heterozygous patients feel almost healthy; they find anemia and morphological changes in red blood cells only under conditions of hypoxia (climbing mountains, heavy physical activity, flying on airplanes, etc.). However, hemolytic crisis and they may end fatally.
Thus, the clinic of sickle cell anemia is characterized by polysymptom: yellowness of the skin, hypoxic syndrome, hepatosplenomegaly, skeletal deformity, repeated organ thrombosis; of hematological symptoms: regenerative anemia, sickle cell red blood cells detected by special tests, hyperbilirubinemia due to the free fraction. A person’s belonging to a certain ethnic group gives reason to suspect this disease and begin a targeted examination to confirm or exclude this anemia.
Sickle cell crisis includes the following; thrombotic crisis (vaso-occlusive infarction); hemolytic crisis, characterized by a sharp increase in hemolysis and accompanied by a catastrophic drop in hematocrit with the simultaneous development of jaundice; aplastic crisis, often triggered by an intercurrent infection that causes bone marrow depression or folic acid deficiency with a life-threatening decrease in hematocrit and the number of reticulocytes in peripheral blood; sequestration crisis (an unusual sign of sickle cell anemia in children), clinically manifested by a sudden painful increase in the liver and spleen with concomitant pancytopenia.
A thrombotic sickle-cell crisis, observed, of course, more often than hemolytic or aplastic, entails a number of progressive pathological changes, the culmination of which is a clinical condition with uncurable pain, prostration and anxiety of the patient. Initially, gnawing pain can occur locally – in the tibia, in the periarticular areas, in the back, stomach or chest. Paroxysmal pain can last only a few hours, but subsequently it resumes with more force and is more prolonged. The described state of the impending crisis, apparently, can occur in patients with sickle cell anemia, experiencing indefinite discomfort with the scarcity of objective research data (examination) and the presence of symptoms that initially appear to be easily eliminated analgesics and intravenous fluids. Any experienced doctor has undoubtedly seen such patients who repeatedly seek help for a 48-72-hour period before the culmination of growing pains requiring hospitalization. The diagnosis of thrombotic crisis is made on the basis of clinical data (before laboratory confirmation); the significance of recent reports of an increase in blood activity of alpha-hydroxybutyrate dehydrogenase in such patients has not yet been proven. An increase in hemolytic activity can be judged (with the availability of initial data) by an increase in the number of reticulocytes in peripheral blood and by a decrease in hematocrit. Subsequent laboratory tests are aimed at identifying triggers, including pneumonia, urinary infection, pharyngitis, acidosis, and dehydration.
The basis of therapy for vaso-occlusive crisis remains anesthesia, rehydration, acidosis correction and treatment of infection. An additional introduction of oxygen is possible, however, it slightly affects the oxygen tension in the stagnated microvasculature. Red blood cell transfusion is reserved for patients with significant anemia (hematocrit less than 17-18%), since free transfusion with hematocrit more than 30% can increase blood viscosity and increase red blood cell aggregation with an increase in microcirculation. Continuous additional administration of folic acid is shown, taking into account the increased production of red blood cells with a shortened life span. The clinical feasibility of drugs that suppress the formation of sickle cell erythrocytes (urea, sodium cyanate) has not been proven.
Diagnosis of Sickle Cell Anemia
The diagnosis of sickle cell anemia is based on an analysis of the physical properties of hemoglobin. The first and oldest method of such an analysis is the study of the so-called “Wet smear”. When a blood smear is wetted with sodium metabisulfite, red blood cells give oxygen and under the microscope you can see a characteristic change in their shape. For greater accuracy, the study is repeated after 24 hours. Another, more common method is based on the detection of hemoglobin of sickle cells by its reduced solubility in some buffer solutions, which is determined by the turbidity of the solution containing such hemoglobin. The widespread use of this method is associated with the ability to quickly obtain results (after 10-15 minutes).
Unfortunately, these methods do not distinguish between a heterozygous state and a homozygous one. Currently, this can only be done using hemoglobin electrophoresis, i.e. analysis of its mobility in an electric field. Without such an analysis, neither accurate diagnosis nor reliable counseling is possible, but for mass examinations it is too expensive and takes a lot of time.
Sickle Cell Anemia Treatment
Sickle cell anemia is an incurable disease today. However, its identification is extremely important for the proper treatment of other diseases in these patients, as well as for providing them with adequate surgical and obstetric care. Proper management of patients with chronic sickle cell anemia helps prevent severe exacerbations (crises) and prolong life.
In some cases, the development of a severe crisis can be prevented by the rapid administration of antibiotics in its early stages (to stop the infection) and by infusion of liquid (hydration). Oxygen, painkillers, intravenous fluids and antibiotics are usually used to treat a crisis. Sometimes you have to resort to a red blood cell transfusion, as well as apply many other drugs, such as anticonvulsants, to relieve individual symptoms.
Sickle Cell Anemia Prevention
Sickle cell anemia cannot be prevented. However, couples with a sign of sickle cell disease are advised to seek genetic counseling.
How can sickle cell anemia be reduced?
The effects of sickle cell anemia can be effectively reduced by establishing a strategic balance between disease management and prevention programs.
Sickle cell anemia is incurable, but its effects can be alleviated by the following measures:
- High fluid intake
- Healthy eating
- Folic Acid Supplements
- Drug treatment of pain
- Treatment of infections with antibiotics
The most cost-effective strategy to reduce the burden of sickle cell anemia is to supplement the treatment of the disease with preventative measures. Low-cost and reliable blood tests can identify couples at risk of having children affected by this disease. Such genetic screening is especially appropriate before marriage or the decision to have a baby and allows spouses to discuss issues related to their family’s health. Subsequent genetic counseling informs carriers of the characteristic genes about the risks that can be passed on to their children, and about the necessary treatment if the child is affected by sickle cell anemia. Antenatal screening for genetic diseases raises specific ethical, legal, and social issues that need to be addressed.