
What is Thalassemia?
Under the thalassemia understand a group of hereditary diseases, manifested by a violation of the synthesis of any of the globin chains. With this form of pathology, hypochromic anemia is noted with a normal or elevated serum iron content.
The disease in question was first described by American pediatricians in 1925. They observed 5 children from families of Italian immigrants. In children, signs of severe hypochromic anemia, a significant increase in the liver and spleen, and bone changes were found. After the publication of the message, the work of Italian authors appeared, describing a similar, but much milder form of the course of thalassemia, in which persons suffering from this pathology survived to adulthood.
The term “thalassemia” was proposed in 1936. Cooley disease was called the thalassemia major (thalassemia major), considering it as a homozygous form of hereditary pathology. For the first time, the idea that thalassemia is the result of impaired synthesis of globin chains was independently expressed by several scientists. Thalassemia, in which the synthesis of the globin beta chain is impaired, is called beta thalassemia; with a-thalassemia, the synthesis of a-chain is impaired. Cases of y-, S-, 8-thalassemia with impaired synthesis of the corresponding globin chains are also described. Beta-thalassemia is more common.
Pathogenesis during Thalassemia
In thalassemia, one of the globin chains is synthesized in small quantities or not synthesized at all. Normally, the synthesis of globin chains is balanced. The number of a– and non-a-chains is the same, and there are no normal free globin chains. Disturbed synthesis of one of them leads to an imbalance. The chain, which is produced in excess, aggregates and is deposited in red blood cells. A large part of the clinical manifestations of thalassemia is associated with this.
With a-thalassemia, in most cases, the chromosome region of the structural genes responsible for the synthesis of the a-chain is lost, it is encoded not by one, but by most people by two pairs of genes located on chromosome 11. The absence of the a-chain in the fetus leads to the development of dropsy and intrauterine death. Deletion in one of the 4 genes encoding the a chain causes a slight deficiency of the a chain; a deletion in 2 genes is a more pronounced deficiency, but the clinical manifestations of the disease largely depend on which 2 genes do not function – on the same chromosome or in different ones. The value of 2 pairs of genes responsible for the synthesis of the a chain is not the same: one pair is the main, the other is the secondary. The clinical picture also depends on in which 2 of the 4 genes the mutation occurred. If 3 genes are absent, then patients have hemoglobinopathy N. Hemoglobin N consists of 4 beta chains. It is very unstable, aggregates, and severe hemolytic anemia develops.
The beta thalassemia mechanism is much more complex. The synthesis of the beta chain is impaired in a number of different diseases: beta-thalassemia, hereditary persistence of fetal hemoglobin, hemoglobinopathy Lepore. However, with these diseases, the disorders are not the same. The gene encoding the synthesis of the beta chain is located on chromosome 16. On the same chromosome, the genes responsible for the synthesis of y and 8 chains are located next to it. In beta-thalassemia, when the beta chain is not produced at all, gene deletions were not detected. With thalassemia, there are signs of RNA instability. The main reason for some cases of beta-thalassemia is a violation of splicing (the process of “stitching” coding sections). By splicing, we understand the changes that mRNA undergoes along the path from the nucleus, where it is synthesized, into the cytoplasm. Primary mRNA transcripts are arranged so that the regions encoding the protein alternate with the regions that do not encode it. These non-coding regions are normally excised from the RNA molecule, and the coding parts of the molecule are connected to each other. Impaired splicing can lead to impaired mRNA stability.
Deletion of the gene responsible for the synthesis of the 8-chain was detected in other forms of pathology. This is a beta, 8-thalassemia. Deletion of the 0– and 8-chain gene was detected during hereditary persistence of fetal hemoglobin. This is a kind of disease with or without very mild anemia, when the content of fetal hemoglobin is greatly increased (up to 95-98%). Unlike homozygous beta-thalassemia, in which sometimes the same high content of fetal hemoglobin is found, with hereditary persistence of fetal hemoglobin there is no imbalance in the synthesis of chains. Lepore hemoglobinopathy is a consequence of a deletion in the genes responsible for the synthesis of 8 and y chains.
Symptoms of Thalassemia
The basis of the clinical manifestations of thalassemia focuses on the excess amount of the globin chain. So, with beta-thalassemia, due to a violation of the synthesis of the beta chain, there are many free a-chains. Excessive synthesis of the a-chain is the main cause of ineffective hematopoiesis in beta-thalassemia. Red blood cells die in the bone marrow. The death of red blood cells and, to a lesser extent, reticulocytes and peripheral blood red blood cells in the spleen leads to severe anemia. In this case, foci of red blood formation can form in the spleen and in the liver. Intense hematopoiesis in the bones can lead to distortion, severe hypoxia – to impair the development of the child. There is a correlation between the depth of anemia and excess a-chain in beta-thalassemia. With a higher content of fetal hemoglobin, cells are destroyed less. This is due to the fact that excess a-chains are not in a free, but in a state associated with the y-chains in the form of hemoglobin F. Some patients with homozygous thalassemia have no severe signs of the disease. Their anemia is not so deep, they can live without constant blood transfusions, and clinically the disease is defined not as a large thalassemia, but as an intermediate thalassemia. There are patients with more severe heterozygous beta-thalassemia. In these patients, inclusions of excess a-chains precipitated are often found. The severity of heterozygous thalassemia in these patients is due to the insufficient ability of cells to free themselves from excess a-chains. Typically, with heterozygous thalassemia, there is a compensatory increase in the utilization of excess a-chains.
With a-thalassemia, in the absence of synthesis of the a-chain and, therefore, hemoglobins A, A2, and F, dropsy of the fetus develops, which leads to its death. Excess beta chains in thalassemia can form hemoglobin, consisting of 4 beta chains, hemoglobin N. Cells containing hemoglobin N are very easily removed from the circulation by the spleen. Anemia in hemoglobinopathy H is caused by both hemolysis of peripheral red blood cells and a violation of globin synthesis.
Homozygous Beta Thalassemia
With homozygous beta-thalassemia, which was described in 1925, clinical manifestations are noted towards the end of the first or, less commonly, second year of a child’s life. In the first months of life, only mild anemia is detected, and it is far from always clear whether the child inherited the disease from one or both parents.
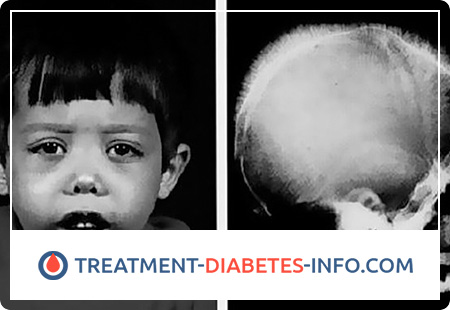
Homozygous beta-thalassemia is accompanied by a significant increase in the spleen, yellowness and a grayish tint of the skin and mucous membranes, marked by pallor. Sharp hyperplasia of the hematopoietic bone marrow leads to a wide variety of skeletal deformations: an almost square skull, a strongly flattened nose bridge, narrowing of the eye cracks are noted.
An X-ray examination reveals an increase in the thickness of the spongy layer of the bones of the cranial vault, as well as transverse striation on the outer plate of the frontal and parietal bones. Children lag behind in physical development, their resistance to various infections is reduced, sexual development is delayed and even disturbed.
The hemoglobin content decreases to 30-50 g / l. The developed erythrocyte hypochromia is detected, the color index, as a rule, is 0.5 or lower. The number of reticulocytes is increased, irritation of the red sprout is much more pronounced than the corresponding reticulocytosis. The content of reticulocytes rises to 2.5-4%. The serum iron content is most often increased, but may not go beyond the upper limit of normal. In the bone marrow, the content of sideroblasts is increased.
An increase in the level of bilirubin due to the indirect fraction is a characteristic feature of homozygous beta-thalassemia.
Due to excessive iron deposition, cirrhosis of the liver, cardiosclerosis, and diabetes mellitus are formed. Underdevelopment of secondary sexual characteristics is also noted.
The severity is distinguished: severe homozygous thalassemia, in which patients die in the first year of life, moderate, in which children live up to 5-8 years, and a lighter form that allows patients to live to adulthood.
The diagnosis of homozygous beta-thalassemia is confirmed by a study of the content of fetal hemoglobin in red blood cells. With homozygous thalassemia, this indicator increases to 20-30.
Heterozygous Beta Thalassemia
Heterozygous beta-thalassemia is the result of the inheritance of the disease from only one of the parents, possibly with an asymptomatic course or with unclear clinical manifestations. Violent clinical manifestations are most rare. Complaints of patients with heterozygous thalassemia are of a general nature and are manifested by weakness, increased fatigue, decreased performance, the most seriously ill patient perceives physical labor. The skin of such patients is pale, a slight ictericity of the skin and sclera is characteristic.
An enlarged spleen is a fairly common symptom of heterozygous beta-thalassemia.
Blood counts for heterozygous thalassemia are extremely diverse. The hemoglobin content may be close to normal. The so-called thalassemia minima is accompanied by hemoglobin indices of 110-120 g / l, or it is reduced to 90-100 g / l (thalassemia minor), rarely the indicator reaches 70 g / l. Anemia is hypochromic. The decrease in the color index can be significant (0.5-0.7). In addition to red blood cell hypochromia, anisocytosis and poikilocytosis, targeting of red blood cells are often detected.
Erythrocyte targeting is also found in iron deficiency anemia, lead intoxication, in persons undergoing splenectomy.
A typical sign of thalassemia is basophilic punctured red blood cells.
The content of reticulocytes with heterozygous thalassemia usually rises to 2-5%. Significant irritation of the red sprout of bone marrow is detected. Often, a red sprout is 1-3 times higher than white. Reduced the number of mature red blood cells containing hemoglobin. The content of iron granules in the bone marrow is increased or, less commonly, normal. The serum iron content in patients with heterozygous thalassemia is normal, less often elevated. Iron reserves determined by the desferal test turn out to be increased.
In 75% of cases of heterozygous beta-thalassemia, an increase in the level of indirect bilirubin is detected.
The diagnosis of heterozygous beta-thalassemia is made on the basis of an increase in the content of the hemoglobin A2 fraction. The content of hemoglobin A2 in heterozygous beta-thalassemia increases to 4.2-8.9% of the total amount of hemoglobin. Approximately half of the patients showed an increase in the content of fetal hemoglobin up to 2.5-7%.
An important diagnostic sign is a similar disease in family members of the patient.
Alpha thalassemia
The synthesis of the a-chain of globin, in contrast to the synthesis of the beta-chain, is controlled by two pairs of unequal genes; in this connection, there are various variants of the course of a-thalassemia.
In the complete absence of the a chain, homozygous dysfunction of all 4 genes, fetal hemoglobin is not synthesized in the fetus, dropsy develops, resulting in death. Impaired function of 1 or 2 genes leads to anemia, usually a mild course. The clinical manifestations of a-thalassemia with damage to 2 genes depend on which genes are affected and almost completely repeat heterozygous beta-thalassemia. Often an enlargement of the spleen is detected, less often – of the liver.
Moderate hypochromic anemia with target erythrocytes and erythrocytes with basophilic puncture, a slight increase in the level of reticulocytes, a slight hyperbilirubinemia and an increase in serum iron, and an increase in the osmotic resistance of red blood cells are determined. A sharp irritation of the red sprout of bone marrow is noted. As a rule, the patient’s blood relatives have the same anemia.
However, unlike beta-thalassemia, the amount of fetal hemoglobin and hemoglobin A2 does not increase with a-thalassemia. α-thalassemia can be diagnosed only in the case of studying the biosynthesis of globin chains in vitro (in vitro).
Alpha-thalassemia is sometimes detected in newborns: a blood test reveals Bart hemoglobin lacking an a chain. This hemoglobin consists of 4 y-chains.
Hemoglobinopathy N is one of the variants of a-thalassemia. It is relatively mild, manifested by an increase in the size of both the spleen and liver, slight jaundice, mainly due to an increase in the fraction of indirect bilirubin, anemia of varying severity, usually the hemoglobin content does not fall below 70–80 g / l. Marked hypochromia of red blood cells, their targeting, basophilic puncture are noted. As with other forms of thalassemia, hemoglobinopathy H shows signs of ineffective hematopoiesis: a sharp irritation of the red bone marrow growth with a slight increase in the level of reticulocytes. Hemoglobinopathy of H differs from other forms of thalassemia by multiple small inclusions in all red blood cells.
Hemoglobinopathy H was first described in 1955 by Rigas et al. These scientists observed 2 patients with hemolytic anemia, in whom, with hemoglobin electrophoresis in an alkaline buffer, an additional fraction was detected moving ahead of hemoglobin A.
Lepore hemoglobinopathy is a form of thalassemia in which globin a-chain synthesis is normal and second-chain synthesis is impaired. Instead of the normal beta chain, a kind of chain is synthesized, consisting of fragments of 0– and 8-chains. The clinical signs of Lepore hemoglobinopathy are extremely similar to those for homozygous beta-thalassemia.
About 70-80% of fetal hemoglobin and about 20% of Lepore hemoglobin are detected. According to clinical and hematological features, the heterozygous form of Lepore hemoglobinopathy cannot be distinguished from heterozygous thalassemia. The hemoglobin content ranges from 90 to 100 g / l, the spleen is often enlarged. In heterozygotes, the content of Lepore hemoglobin is 7-8%, while the content of hemoglobin A2 is reduced and the content of fetal hemoglobin is increased.
Lepore hemoglobinopathies are rare.
Thalassemia Treatment
Treatment of homozygous thalassemia. Since the manifestations of the disease are determined by hypoxia and active hematopoiesis in the bones, where it is usually absent, red blood cells are transfused to the patient from an early age.
Initially, a shock treatment course is applied (for 2-3 weeks, 8-10 transfusions). In this case, the hemoglobin content usually rises to 120-140 g / l. Then transfusions are done less frequently, every 3-4 weeks, at the rate of 20 ml / kg. They try to maintain hemoglobin levels at 90-100 g / l. Massive transfusions with thalassemia not only improve the general condition, but also reduce skeletal changes, enlarge the spleen, improve the development of children, and reduce the incidence of severe infections.
Frequent complications of this therapy are pyrogenic reactions, mainly with the use of whole blood or insufficiently washed red blood cells.
One of the serious complications of transfusion is excessive deposition of iron-containing pigment, which leads to a significant increase in the liver, diabetes mellitus, heart failure. Treatment for homozygous thalassemia involves the mandatory use of desferal to excrete excess iron. The dose of desferal (administered intramuscularly) depends on the number of transfused red blood cells and the patient’s age: for young children – 10 mg / kg, for adolescents – 500 mg / day. Large doses are also acceptable – 1 g / day. It is recommended to combine desferal with 200-500 mg of ascorbic acid inside.
A significant increase in the spleen, the addition of leukopenia, thrombocytopenia to the signs of hypochromic anemia makes us think of splenectomy.
Treatment of homozygous thalassemia with these methods improves the general condition of patients, increases their life expectancy.
Currently, bone marrow transplantation is widely used for patients with thalassemia, as a result of which, in most cases, persistent improvement is achieved.
Treatment of heterozygous beta-thalassemia in most cases is not required. Patients feel satisfactory. The hemoglobin content ranges from 90 to 110 g / l. With heterozygous beta-thalassemia, blood transfusions are not required. Removal of the spleen is extremely rare and only with a very large spleen.
Desferal should be used for heterozygous beta-thalassemia, when there is a high amount of serum iron and iron in the urine after administration of 500 mg of desferal. However, desferal only prevents siderosis (a human disease caused by the deposition of dust containing iron in the lungs), without leading to an increase in hemoglobin levels.
With a decrease in hemoglobin due to infectious diseases, during pregnancy, folic acid should be used at 0.005 g 1-2 times a day, since with ineffective hematopoiesis associated with thalassemia, a significant amount of folic acid is consumed.
With heterozygous thalassemia, iron preparations are contraindicated, since there is always some excess of iron without the clinical manifestations of hemosiderosis. However, patients taking iron supplements feel significantly worse. The most dangerous for thalassemia is the parenteral administration of iron preparations: patients who are in satisfactory condition may die from severe hemosiderosis, in particular from circulatory failure associated with myocardial siderosis.
The treatment of a-thalassemia is practically no different from the treatment of heterozygous thalassemia, with the exception of hemoglobinopathy N.
With hemoglobinopathy N, due to the instability of this hemoglobin and its precipitation, there are signs of not only ineffective hematopoiesis, but also pronounced destruction of peripheral blood red blood cells. This destruction occurs mainly in the spleen, as a result of which it increases. The main treatment for hemoglobinopathy H in severe anemia is the removal of the spleen.